
Reactions of hydroxyl radicals with benzoic acid and benzoate†

Abstract
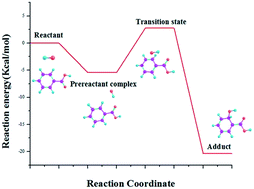
Density functional theory was used to study the mechanism and kinetics of benzoic acid with hydroxyl radicals in both gas and aqueous phases as well as benzoate with hydroxyl radicals in the aqueous phase at the M06-2X/6-311+G(d,p) level of theory. The results show that all reaction pathways involved the formation of pre-reactive complexes which in turn alter reaction energy barriers. The reaction rate constants, calculated based on classical transitional theory, followed the order of meta addition > para addition > ortho addition for the reaction of benzoic acid and hydroxyl radicals in both gas and aqueous media. The energy barrier analysis reveals that the ortho adducts were also less vulnerable to subsequent reaction. In addition, the rate constants for the addition reactions were highest for benzoate in the aqueous phase, followed by benzoic acid in the aqueous phase, then by benzoic acid in the gas phase, consistent with electrostatic potential analysis. However, the rate constants of hydrogen abstraction in the aqueous phase were much lower than that in the gas phase and thus, gas phase reactions are preferred. The incorporation of one explicit water molecule, for addition reactions between benzoic acid and hydroxyl radicals, lowered reaction rates in the aqueous phase by increasing the bond length between the oxygen and reacting carbon in the benzene ring.
 Please wait while we load your content...
Please wait while we load your content...

