
First-principles study of Zr–N crystalline phases: phase stability, electronic and mechanical properties†
 *ab
*ab
Abstract
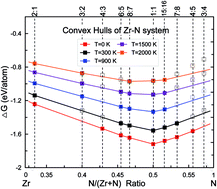
Using a variable-composition ab initio evolutionary algorithm, we investigate stability of various Zr–N compounds. Besides the known ZrN and Zr3N4, new candidate structures with Zr : N ratios of 2 : 1, 4 : 3, 6 : 5, 8 : 7, 15 : 16, 7 : 8 and 4 : 5 are found to be ground-state configurations, while Zr3N2 has a very slightly higher energy. Besides Zr2N, the newly discovered ZrxNy compounds adopt rocksalt structures with ordered nitrogen or zirconium vacancies. The electronic and mechanical properties of the zirconium nitrides are further studied in order to understand their composition–structure–property relationships. Our results show that bulk and shear moduli monotonically increase with decreasing vacancy content. The mechanical enhancement can be attributed to the occurrence of more Zr–N covalent bonds and weakening of the ductile Zr–Zr metallic bonds. These simulations could provide additional insight into the vacancy-ordered rocksalt phases that are not readily apparent from experiments.
 Please wait while we load your content...
Please wait while we load your content...

