
DFT insights into the electronic properties and adsorption of NO2 on metal-doped carbon nanotubes for gas sensing applications†

Abstract
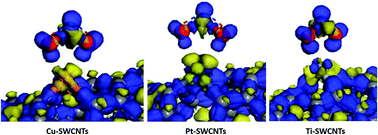
The gas sensing process involves complicated and precise adsorption mechanisms, and the selection of the right material is mainly based on “guess-and-check” procedures, which necessitates the search for a more systematic approach to discover optimum materials with required specifications. Herein, we report the use of ab initio first-principles methods to expedite the process of finding the material with the highest potential compared to in-lab trial and error. We focused on the NO2 adsorption mechanism of Cu-, Pt- and Ti-doped single-walled carbon nanotubes (SWCNTs). Modelling the isolated NO2 molecule indicated the formation of a band gap between the 3π* HOMO and 5σ* LUMO levels, which were found to be the electronic states primarily involved in the bonding process. Metal doping decreased the system stability and altered the SWCNT structure. NO2 exposure caused the transfer of electrons to the gas molecules, thereby enhancing the p-type conductivity of the M-SWCNT structure. The highest charge transfer to the NO2 molecule was observed for Ti-SWCNTs (0.456 eV), while the lowest was found for Cu-SWCNTs (0.351 eV). Ti-SWCNTs exhibited the highest stability and sensitivity as a potential NO2 gas sensing material out of the 3 investigated metal dopants.
 Please wait while we load your content...
Please wait while we load your content...

