
Changing the gap type of solid state boric acid by heating: a dispersion-corrected density functional study of α-, β-, and γ-metaboric acid polymorphs†
 *c
and
*c
and
Abstract
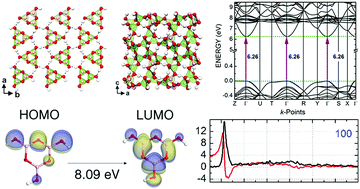
The metaboric acid α-, β-, and γ-polymorphs were investigated using density functional theory (DFT) calculations by employing the generalized gradient approximation improved using the Tkatchenko–Scheffler scheme to take into account dispersive interactions. The structural, electronic, and optical properties of these polymorphs were thus obtained, with unit cell deviations Δa, Δb, and Δc (in comparison with X-ray data) as small as −0.08, −0.09, and −0.06 Å for α-(BOH)3O3, 0.00, 0.06, and −0.09 Å for β-(BOH)3O3, and 0.01, 0.01, and 0.01 Å for γ-(BOH)3O3. The layered α-MA polymorph is predicted by our simulations to have a direct gap of 6.26 eV, which is close to the values found recently for the boric acid triclinic (H3BO3-2A, 6.25 eV) and trigonal (H3BO3-3T, 6.28 eV) indirect gap systems. As the α-MA structure can be obtained by heating up the H3BO3-2A crystal above 100 °C, we have an interesting result that a change in temperature can modify the nature of the band gap of boric acid in the solid state from indirect to direct. van der Waals interactions between the metaboric acid α-(BOH)3O3 planes (distance of about 3.1 ± 0.01 Å) are a relevant aspect determining its energy gap, while its direct or indirect character depends on the crystal structure and molecular components. On the other hand, the polymeric monoclinic β-(BOH)3O3 has an indirect gap of 6.56 eV, while the covalent cubic γ-(BOH)3O3 has a direct gap of 7.28 eV. Finally, the complex dielectric function and optical absorption considering polarized light along the 001, 010, 100 crystal planes and polycrystalline samples (POLY) of the three metaboric acid polymorphs were obtained.
 Please wait while we load your content...
Please wait while we load your content...

