
An innovative gas sensor system designed from a sensitive nanostructured ZnO for the selective detection of SOx molecules: a density functional theory study
 *abc
and
*abc
and
Abstract
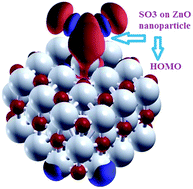
The adsorption behaviors of SOx molecules on pristine and N-doped ZnO nanoparticles were investigated using density functional theory calculations (DFT). The results suggest an improved adsorption ability for N-doped nanoparticles over undoped nanoparticles. Various adsorption geometries and sites were considered in detail. In all adsorption sites, the SOx molecule tends to form a bridge geometry with the ZnO nanoparticle, giving rise to multiple contacting points between the nanoparticle and SOx molecule. SOx adsorption on the N-doped ZnO nanoparticle is found to be more favorable in energy than the adsorption on the undoped nanoparticle, suggesting that the N-doped nanoparticles have a higher sensing capability than the pristine nanoparticles. After adsorption, the S–O bonds of the adsorbed SOx molecule were stretched, which can be attributed to the transfer of electron density from the S–O bonds to the newly formed bonds between the ZnO and SOx molecule. The charge analysis based on the natural bond orbital (NBO) method reveals a considerable charge transfer from the adsorbed SOx molecule to the ZnO nanoparticle, indicating the donor properties of SOx molecules during the adsorption process. By analyzing the projected density of states, it was found that chemical bonds were formed between the interacting atoms at the interface region. The results also indicate that the electronic densities in the highest occupied molecular orbitals (HOMOs) were mainly distributed over the SOx molecules, whereas the lowest unoccupied molecular orbitals (LUMOs) were dominant at the ZnO nanoparticle. Our DFT calculations shed light on the improved adsorption behaviors of N-doped nanoparticles as innovative gas sensors for SOx detection in the environment.
 Please wait while we load your content...
Please wait while we load your content...

