
Protein–ligand (un)binding kinetics as a new paradigm for drug discovery at the crossroad between experiments and modelling†
 *b
*b
Abstract
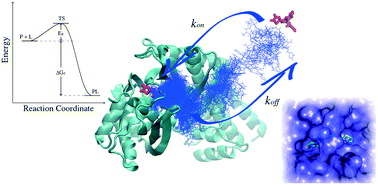
In the last three decades, protein and nucleic acid structure determination and comprehension of the mechanisms, leading to their physiological and pathological functions, have become a cornerstone of biomedical sciences. A deep understanding of the principles governing the fates of cells and tissue at the molecular level has been gained over the years, offering a solid basis for the rational design of drugs aimed at the pharmacological treatment of numerous diseases. Historically, affinity indicators (i.e. Kd and IC50/EC50) have been assumed to be valid indicators of the in vivo efficacy of a drug. However, recent studies pointed out that the kinetics of the drug–receptor binding process could be as important or even more important than affinity in determining the drug efficacy. This eventually led to a growing interest in the characterisation and prediction of the rate constants of protein–ligand association and dissociation. For instance, a drug with a longer residence time can kinetically select a given receptor over another, even if the affinity for both receptors is comparable, thus increasing its therapeutic index. Therefore, understanding the molecular features underlying binding and unbinding processes is of central interest towards the rational control of drug binding kinetics. In this review, we report the theoretical framework behind protein–ligand association and highlight the latest advances in the experimental and computational approaches exploited to investigate the binding kinetics.
 Please wait while we load your content...
Please wait while we load your content...