
Requirements for reversible extra-capacity in Li-rich layered oxides for Li-ion batteries†
 ab
and
M.-L.
Doublet
*ab
ab
and
M.-L.
Doublet
*ab
Abstract
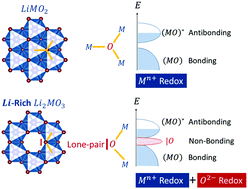
The structural stability and the redox mechanism of Li-rich layered oxides (LLOs) are two very important aspects for high energy density. The former is related to the irreversible loss of lattice oxygen and capacity fading during cycling, while the latter determines the overall capacity of the materials. This paper aims at clarifying the factors governing the structural stability, the extra capacity and the redox mechanism of LLOs upon Li-removal. The results show that the structural stability against oxygen vacancy formation is improved with increasing M–O covalency, while it decreases with increasing d-shell electron number and with electrochemical extraction of lithium from the lattice. The redox mechanism of Li2−xMO3 electrodes formed by 3d metals or by heavier metals with a d0 electronic configuration is related to the electron depletion from the oxygen lone-pairs (localized non-bonding O(2p) states) leading to an irreversible anionic redox ending with the reductive elimination of O2 upon cycling. For these phases, long-term cycling is predicted to be very unlikely due to the irreversible loss of lattice oxygen upon charging. For the electrodes formed by 4d and 5d metals with intermediate dn electronic configurations, reversible cationic and anionic redox activities are predicted, therefore enabling reversible extra-capacities. The very different redox mechanisms exhibited by Li2−xMO3 electrodes are then linked to the delicate balance between the Coulomb repulsions (U term) and the M–O bond covalency (Δ term) through the general description of charge-transfer vs. Mott–Hubbard insulators. The present findings will provide a uniform guideline for tuning the band structures of Li2MO3 phases and thus activating desired redox mechanisms, being beneficial for the design of high-energy density electrode materials for Li-ion battery applications.
 Please wait while we load your content...
Please wait while we load your content...

