
The ligand effect on the oxidative addition of dioxygen to gold(i)–hydride complexes†

Abstract
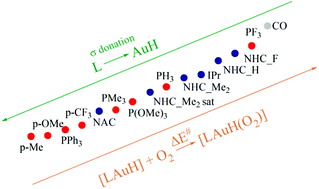
The ligand effect on the recently uncovered feasible oxidative addition reaction of O2 on [LAuH] complexes has been investigated for a series of fifteen ligands. The activation barriers of this spin-forbidden reaction have been estimated at the minimum energy crossing points (MECP, relativistic scalar level) between the adiabatic triplet (reactants spin state) and singlet (product spin state) potential energy surfaces (PES) and calculated at the transition states by including Spin–Orbit Coupling (SOC) effects, as applied for the mechanistic study of this reaction in a previous study by us [Chem. Sci., 2016, 7, 7034–7039]. We find a sizeable effect of the ligand on the activation barriers, and some of the stronger electron donating phosphines are predicted to induce the highest catalyst efficiency. The inclusion of SOC effects lowers the activation barriers by about 3 kcal mol−1 systematically with respect to the MECP values independently of the ligand type. We used the Charge-Displacement (CD) analysis to quantify the net electron charge donation from the ligand L towards the metallic fragment AuH in the [LAuH] series, and surprisingly only a poor correlation was found between the net electron donor character of L and the activation barriers. Application of the CD-NOCV (Natural Orbitals for Chemical Valence) approach, which allows the quantification of the Dewar–Chatt–Duncanson (DCD) L–AuH bond components, suggests that the ligand effect on the activation barriers is not easily predictable on the basis of solely the electronic properties of the ligand and depends significantly on the ligand nature or carbene or phosphine type. We show that for both phosphine and carbene ligand subsets, however, the σ donation component of the L–AuH bond quantitatively accounts for the ligand effect on the activation energy barriers (a larger σ-donor capability of L correlates with a smaller activation barrier), whereas the π back-donation, strongly affected by geometrical rearrangement, is a poor reactivity descriptor (π acceptor properties of the ligand L in the linear [LAuH] complexes are not transferable to the trigonal [LAuH(O2)] transition state structures).
 Please wait while we load your content...
Please wait while we load your content...

