
The effect of auxiliary ligand on the mechanism and reactivity: DFT study on H2 activation by Lewis acid–transition metal complex (tris(phosphino)borane)Fe(L)†
 ‡a
Zhuofeng
Ke
*a
‡a
Zhuofeng
Ke
*a
Abstract
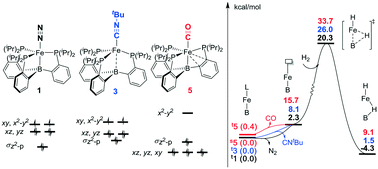
Lewis acid–transition metal (LA–TM) complexes have been emerging as a novel type of bifunctional catalyst for H2 activation. The crucial role of the auxiliary ligand in the mechanism and reactivity of H2 activation were theoretically studied with (tris(phosphino)borane)Fe(L) (L = N2, CNtBu, and CO) as model LA–TM complexes. The axial auxiliary ligand is found to play an important role in the complex electronic structures, via significantly tuning the energy levels of dxz, dyz, and 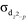 of the Fe–B bond. We systematically evaluated both the ligand dissociative mechanism and the associative mechanism with the binding auxiliary ligand. In the ligand dissociative mechanism, the reaction starts with the dissociation of the axial ligand. Then, H2 coordinates to the iron center either at the axial position or on the equatorial plane. The axial coordinated H2 cleaves via oxidative addition to a metastable octahedral dihydride intermediate, which further isomerizes to a more stable five-coordinated trigonal bipyramidal intermediate with a bridging hydride stabilized by the iron center and the Lewis center, boron. On the other hand, the equatorial coordinated H2 is cleaved by the cooperation of the Fe–B bond, directly to the trigonal bipyramidal dihydride intermediate. In comparison, in the ligand associative mechanism, the H2 molecule splits up upon approaching the iron center via an octahedral transition state, with the auxiliary ligand remaining at the axial position. Our results suggest that the triplet state axial reaction pathway of the L-dissociative mechanism is the most favorable one for H2 activation. The isomerization of hydride instead of H2 cleavage may be the rate-determining step. H2 activation occurs homolytically on the metal center without LA assistance, which is significantly different from other tetradentate LA–TM systems that activate H2 in a synergetic heterolytic mode. The obtained tendency of the dissociation free energies for (tris(phosphino)borane)Fe(L) (L = N2, CNtBu, and CO) (2.3, 8.1, and 15.7 kcal mol−1, respectively) and the activation free energies of the rate-determining step (20.3, 26.0, and 33.7 kcal mol−1, respectively) explains well their activity trend. The extraordinary effect of the auxiliary ligand on the mechanism and reactivity should provide new information for future development of LA–TM bifunctional catalysts.
of the Fe–B bond. We systematically evaluated both the ligand dissociative mechanism and the associative mechanism with the binding auxiliary ligand. In the ligand dissociative mechanism, the reaction starts with the dissociation of the axial ligand. Then, H2 coordinates to the iron center either at the axial position or on the equatorial plane. The axial coordinated H2 cleaves via oxidative addition to a metastable octahedral dihydride intermediate, which further isomerizes to a more stable five-coordinated trigonal bipyramidal intermediate with a bridging hydride stabilized by the iron center and the Lewis center, boron. On the other hand, the equatorial coordinated H2 is cleaved by the cooperation of the Fe–B bond, directly to the trigonal bipyramidal dihydride intermediate. In comparison, in the ligand associative mechanism, the H2 molecule splits up upon approaching the iron center via an octahedral transition state, with the auxiliary ligand remaining at the axial position. Our results suggest that the triplet state axial reaction pathway of the L-dissociative mechanism is the most favorable one for H2 activation. The isomerization of hydride instead of H2 cleavage may be the rate-determining step. H2 activation occurs homolytically on the metal center without LA assistance, which is significantly different from other tetradentate LA–TM systems that activate H2 in a synergetic heterolytic mode. The obtained tendency of the dissociation free energies for (tris(phosphino)borane)Fe(L) (L = N2, CNtBu, and CO) (2.3, 8.1, and 15.7 kcal mol−1, respectively) and the activation free energies of the rate-determining step (20.3, 26.0, and 33.7 kcal mol−1, respectively) explains well their activity trend. The extraordinary effect of the auxiliary ligand on the mechanism and reactivity should provide new information for future development of LA–TM bifunctional catalysts.
 Please wait while we load your content...
Please wait while we load your content...

