
The complete reaction mechanism of H2S desulfurization on an anatase TiO2 (001) surface: a density functional theory investigation†

Abstract
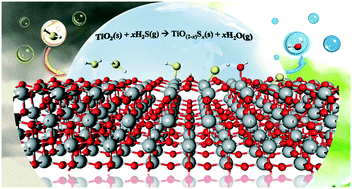
The complete reaction mechanism of H2S desulfurization on anatase TiO2 (001) surface was elucidated using the plane-wave based density functional theory (DFT) method. The reaction starts from the dissociative adsorption of H2S on the TiO2 surface. Subsequently, two competitive routes, H2O and H2 formation, were investigated. The activation barriers for H2O formation range from 11 to 13 kcal mol−1, whereas those for H2 formation are extremely high in the range of 67–87 kcal mol−1. On the basis of the activation energy barriers, the results indicate that the anatase TiO2 (001) is very active for H2S desulfurization to produce H2O, resulting in S-substitution at the O2c sites on the TiO2 (001) surface. Electronic charge analyses indicate that S-doping onto the TiO2 surface can enhance the photocatalytic activity of TiO2 by reducing its band gap. In addition, by comparison with other metal oxide catalysts, such as TiO2 (101), CeO2 (111), CeO2 (101), ZnO (1010) and α-Fe2O3 (0001), we found that TiO2 (001) is the most promising catalyst for H2S desulfurization.
 Please wait while we load your content...
Please wait while we load your content...

