
Quantum and quasiclassical trajectory studies of rotational relaxation in Ar–N2+ collisions

Abstract
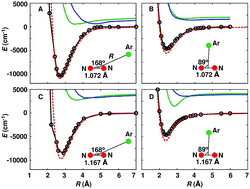
The collision of N2+ with Ar is studied using quantum and classical methods. The dynamics was followed on a new potential energy surface based on ab initio energies computed at the UCCSD(T)-F12a/aug-cc-pVTZ level, using the correct analytical long range behaviour and a reproducing kernel representation. Comparison with multi-reference MRCI+Q calculations establish that UCCSD(T)-F12a is a sufficiently high level of theory for this problem. Results from quantum close coupling and quasiclassical trajectory calculations agree favourably with each other and the rates for inelastic collisions are lower than those from Langevin theory. This differs from previous calculations on a zero point-corrected potential energy surface (PES) and indicates that such corrections, although potentially useful, should not be applied in the present case. Despite the rather large differences between the potential energy surfaces, the computed rates are within one order of magnitude of one another which suggests that the quality of the PES is not the main reason for the remaining disagreement between computation and experiment. Also, the fraction of inelastic rotational collisions exceeds 20% in all cases irrespective of whether quantum or classical dynamics is used. Previous experimental rate coefficients for N2+(ν = 0, j = 6) colliding with Ar suggest that the rotational quantum number is largely conserved. This can not be confirmed from any of the simulations and calls for new single molecule experiments.
 Please wait while we load your content...
Please wait while we load your content...

