
First-principles calculations of oxygen interstitials in corundum: a site symmetry approach†
 *a
Alexander
Platonenko,
b
Denis
Gryaznov,
b
Yuri F.
Zhukovskii
b
and
Eugene A.
Kotomin
bc
*a
Alexander
Platonenko,
b
Denis
Gryaznov,
b
Yuri F.
Zhukovskii
b
and
Eugene A.
Kotomin
bc
Abstract
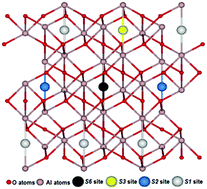
Using site symmetry analysis, four possible positions of interstitial oxygen atoms in the α-Al2O3 hexagonal structure have been identified and studied. First principles hybrid functional calculations of the relevant atomic and electronic structures for interstitial Oi atom insertion in these positions reveal differences in energies of ∼1.5 eV. This approach allows us to get the lowest energy configuration, avoiding time-consuming calculations. It is shown that the triplet oxygen atom is barrierless displaced towards the nearest regular oxygen ion, forming a singlet dumbbell (split interstitial) configuration with an energy gain of ∼2.5 eV. The charge and spatial structure of the dumbbell is discussed. Our results are important, in particular, for understanding the radiation properties and stability of α-Al2O3 and other oxide crystals.
 Please wait while we load your content...
Please wait while we load your content...

