
Ultrafast charge dynamics in glycine induced by attosecond pulses

Abstract
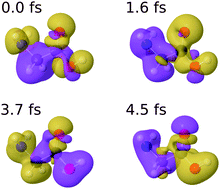
The combination of attosecond pump–probe techniques with mass spectrometry methods has recently led to the first experimental demonstration of ultrafast charge dynamics in a biomolecule, the amino acid phenylalanine [Calegari et al., Science, 2014, 346, 336]. Using an extension of the static-exchange density functional theory (DFT) method, the observed dynamics was explained as resulting from the coherent superposition of ionic states produced by the broadband attosecond pulse. Here, we have used the static-exchange DFT method to investigate charge migration induced by attosecond pulses in the glycine molecule. We show that the observed dynamics follows patterns similar to those previously found in phenylalanine, namely that charge fluctuations occur all over the molecule and that they can be explained in terms of a few typical frequencies of the system. We have checked the validity of our approach by explicitly comparing with the photoelectron spectra obtained in synchrotron radiation experiments and with the charge dynamics that follows the removal of an electron from a given molecular orbital, for which fully correlated ab initio results are available in the literature. From this comparison, we conclude that our method provides an accurate description of both the coherent superposition of cationic states generated by the attosecond pulse and its subsequent time evolution. Hence, we expect that the static-exchange DFT method should perform equally well for other medium-size and large molecules, for which the use of fully correlated ab initio methods is not possible.
- This article is part of the themed collection: XUV/X-ray light and fast ions for ultrafast chemistry
 Please wait while we load your content...
Please wait while we load your content...

