
The role of group III, IV elements in Nb4AC3 MAX phases (A = Al, Si, Ga, Ge) and the unusual anisotropic behavior of the electronic and optical properties†
 ‡b
Weiwei
Sun
§¶*d
‡b
Weiwei
Sun
§¶*d
Abstract
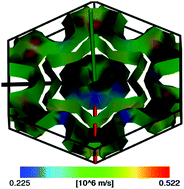
Niobium based Nb4AlC3, Nb4SiC3, Nb4GeC3 and Nb4GaC3 were investigated by means of density functional theory. Together with the known Nb4AlC3, the role of group III, IV elements in various properties of Nb4AC3 (A = Al, Si, Ga, Ge) was systematically investigated, and particularly the bulk moduli, shear moduli, and Young's moduli helped us to approach the ductility. All the studied compounds were found to be mechanically stable, and they also exhibit the metallic nature that results from the Nb-4d states being dominant at the Fermi level. The typical 4d–2p hybridization leads to strong Nb–C covalent bonding and a relatively weaker 4d–3p (4p) hybridization between Nb and A is identified. The latter does perturb the performance of materials. By varying A elements in Nb4AC3, the position and the width of the p states as well as hybridizations are altered, which determine the covalency and the ionicity of the chemical bonds. A high density of states at the Fermi level and the nesting effects in the Fermi surface are identified in Nb4SiC3 and linked to its unusual anisotropic behavior. Furthermore, Nb4GeC3 is predicted to be a very promising candidate solar heating barrier material. Overall, the present work gives insights into the role of A elements in the electronic structure and the physical properties of Nb4AC3 compounds. The tendencies and rules established here will help in the designing of functional ceramic materials with desirable properties.
 Please wait while we load your content...
Please wait while we load your content...

