
Time-dependent vibrational spectral analysis of first principles trajectory of methylamine with wavelet transform

Abstract
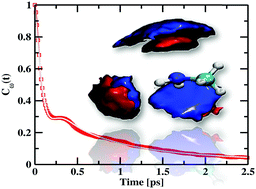
The fluctuation dynamics of amine stretching frequencies, hydrogen bonds, dangling N–D bonds, and the orientation profile of the amine group of methylamine (MA) were investigated under ambient conditions by means of dispersion-corrected density functional theory-based first principles molecular dynamics (FPMD) simulations. Along with the dynamical properties, various equilibrium properties such as radial distribution function, spatial distribution function, combined radial and angular distribution functions and hydrogen bonding were also calculated. The instantaneous stretching frequencies of amine groups were obtained by wavelet transform of the trajectory obtained from FPMD simulations. The frequency–structure correlation reveals that the amine stretching frequency is weakly correlated with the nearest nitrogen–deuterium distance. The frequency–frequency correlation function has a short time scale of around 110 fs and a longer time scale of about 1.15 ps. It was found that the short time scale originates from the underdamped motion of intact hydrogen bonds of MA pairs. However, the long time scale of the vibrational spectral diffusion of N–D modes is determined by the overall dynamics of hydrogen bonds as well as the dangling ND groups and the inertial rotation of the amine group of the molecule.
 Please wait while we load your content...
Please wait while we load your content...

