
Tailoring the structure and thermoelectric properties of BaTiO3via Eu2+ substitution†

Abstract
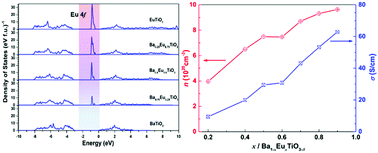
A series of Ba1−xEuxTiO3−δ (0.1 ≤ x ≤ 0.9) phases with ∼40 nm particle size were synthesized via a Pechini method followed by annealing and sintering under a reducing atmosphere. The effects of Eu2+ substitution on the BaTiO3 crystal structure and the thermoelectric transport properties were systematically investigated. According to synchrotron X-ray diffraction data only cubic perovskite structures were observed. On the local scale below about 20 Å (equal to ∼5 unit cells) deviations from the cubic structure model (Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) were detected by evaluation of the pair distribution function (PDF). These deviations cannot be explained by a simple symmetry breaking model like in EuTiO3−δ. The best fit was achieved in the space group Amm2 allowing for a movement of Ti and Ba/Eu along 〈110〉 of the parent unit cell as observed for BaTiO3. Density functional calculations delivered an insight into the electronic structure of Ba1−xEuxTiO3−δ. From the obtained density of states a significant reduction of the band gap by the presence of filled Eu2+ 4f states at the top of the valence band was observed. The physical property measurements revealed that barium–europium titanates exhibit n-type semiconducting behavior and at high temperature the electrical conductivity strongly depended on the Eu2+ content. Activation energies calculated from the electrical conductivity and Seebeck coefficient data indicate that at high temperatures (800 K < T < 1123 K) the conduction mechanism of Ba1−xEuxTiO3−δ (0.1 ≤ x ≤ 0.9) is a polaron hopping when 0 < x ≤ 0.6 and is a thermally activated process when 0.6 < x < 1. Besides, the thermal conductivity increases with increasing Eu2+ concentration. Due to a remarkable improvement of the power factor, Ba0.1Eu0.9TiO3−δ showed a ZT value of 0.24 at 1123 K.
m) were detected by evaluation of the pair distribution function (PDF). These deviations cannot be explained by a simple symmetry breaking model like in EuTiO3−δ. The best fit was achieved in the space group Amm2 allowing for a movement of Ti and Ba/Eu along 〈110〉 of the parent unit cell as observed for BaTiO3. Density functional calculations delivered an insight into the electronic structure of Ba1−xEuxTiO3−δ. From the obtained density of states a significant reduction of the band gap by the presence of filled Eu2+ 4f states at the top of the valence band was observed. The physical property measurements revealed that barium–europium titanates exhibit n-type semiconducting behavior and at high temperature the electrical conductivity strongly depended on the Eu2+ content. Activation energies calculated from the electrical conductivity and Seebeck coefficient data indicate that at high temperatures (800 K < T < 1123 K) the conduction mechanism of Ba1−xEuxTiO3−δ (0.1 ≤ x ≤ 0.9) is a polaron hopping when 0 < x ≤ 0.6 and is a thermally activated process when 0.6 < x < 1. Besides, the thermal conductivity increases with increasing Eu2+ concentration. Due to a remarkable improvement of the power factor, Ba0.1Eu0.9TiO3−δ showed a ZT value of 0.24 at 1123 K.
 Please wait while we load your content...
Please wait while we load your content...

