
Structural and energetic study of cation–π–cation interactions in proteins†
 a
Ignacio
Soteras,
b
Josep Lluis
Gelpí,
c
François
Dehez,
d
F. Javier
Luque
b
and
Carles
Curutchet
*a
a
Ignacio
Soteras,
b
Josep Lluis
Gelpí,
c
François
Dehez,
d
F. Javier
Luque
b
and
Carles
Curutchet
*a
Abstract
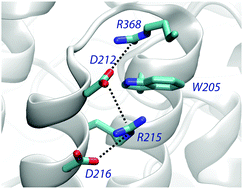
Cation–π interactions of aromatic rings and positively charged groups are among the most important interactions in structural biology. The role and energetic characteristics of these interactions are well established. However, the occurrence of cation–π–cation interactions is an unexpected motif, which raises intriguing questions about its functional role in proteins. We present a statistical analysis of the occurrence, composition and geometrical preferences of cation–π–cation interactions identified in a set of non-redundant protein structures taken from the Protein Data Bank. Our results demonstrate that this structural motif is observed at a small, albeit non-negligible frequency in proteins, and suggest a preference to establish cation–π–cation motifs with Trp, followed by Tyr and Phe. Furthermore, we have found that cation–π–cation interactions tend to be highly conserved, which supports their structural or functional role. Finally, we have performed an energetic analysis of a representative subset of cation–π–cation complexes combining quantum-chemical and continuum solvation calculations. Our results point out that the protein environment can strongly screen the cation–cation repulsion, leading to an attractive interaction in 64% of the complexes analyzed. Together with the high degree of conservation observed, these results suggest a potential stabilizing role in the protein fold, as demonstrated recently for a miniature protein (Craven et al., J. Am. Chem. Soc. 2016, 138, 1543). From a computational point of view, the significant contribution of non-additive three-body terms challenges the suitability of standard additive force fields for describing cation–π–cation motifs in molecular simulations.
 Please wait while we load your content...
Please wait while we load your content...

