
Energy decomposition analysis in an adiabatic picture†
 ‡a
‡a
Abstract
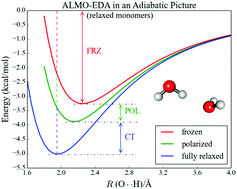
Energy decomposition analysis (EDA) of electronic structure calculations has facilitated quantitative understanding of diverse intermolecular interactions. Nevertheless, such analyses are usually performed at a single geometry and thus decompose a “single-point” interaction energy. As a result, the influence of the physically meaningful EDA components on the molecular structure and other properties are not directly obtained. To address this gap, the absolutely localized molecular orbital (ALMO)-EDA is reformulated in an adiabatic picture, where the frozen, polarization, and charge transfer energy contributions are defined as energy differences between the stationary points on different potential energy surfaces (PESs), which are accessed by geometry optimizations at the frozen, polarized and fully relaxed levels of density functional theory (DFT). Other molecular properties such as vibrational frequencies can thus be obtained at the stationary points on each PES. We apply the adiabatic ALMO-EDA to different configurations of the water dimer, the water–Cl− and water–Mg2+/Ca2+ complexes, metallocenes (Fe2+, Ni2+, Cu2+, Zn2+), and the ammonia–borane complex. This method appears to be very useful for unraveling how physical effects such as polarization and charge transfer modulate changes in molecular properties induced by intermolecular interactions. As an example of the insight obtained, we find that a linear hydrogen bond geometry for the water dimer is preferred even without the presence of polarization and charge transfer, while the red shift in the OH stretch frequency is primarily a charge transfer effect; by contrast, a near-linear geometry for the water–chloride hydrogen bond is achieved only when charge transfer is allowed.
 Please wait while we load your content...
Please wait while we load your content...

