
Thermodynamic stability of stoichiometric LaFeO3 and BiFeO3: a hybrid DFT study†
 *a
*a
Abstract
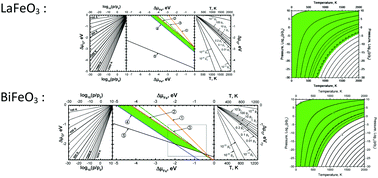
BiFeO3 perovskite attracts great attention due to its multiferroic properties and potential use as a parent material for Bi1−xSrxFeO3−δ and Bi1−xSrxFe1−yCoyO3−δ solid solutions in intermediate temperature cathodes of oxide fuel cells. Another iron-based LaFeO3 perovskite is the end member for well-known solid solutions (La1−xSrxFe1−yCoyO3−δ) used for oxide fuel cells and other electrochemical devices. In this study an ab initio hybrid functional approach was used for the study of the thermodynamic stability of both LaFeO3 and BiFeO3 with respect to decompositions to binary oxides and to elements, as a function of temperature and oxygen pressure. The localized (LCAO) basis sets describing the crystalline electron wave functions were carefully re-optimized within the CRYSTAL09 computer code. The results obtained by considering Fe as an all-electron atom and within the effective core potential technique are compared in detail. Based on our calculations, the phase diagrams were constructed allowing us to predict the stability region of stoichiometric materials in terms of atomic chemical potentials. This permits determining the environmental conditions for the existence of stable BiFeO3 and LaFeO3. These conditions were presented as contour maps of oxygen atoms' chemical potential as a function of temperature and partial pressure of oxygen gas. A similar analysis was also performed using the experimental Gibbs energies of formation. The obtained phase diagrams and contour maps are compared with the calculated ones.
 Please wait while we load your content...
Please wait while we load your content...

