
Conformational structures of jet-cooled acetaminophen–water clusters: a gas phase spectroscopic and computational study†

Abstract
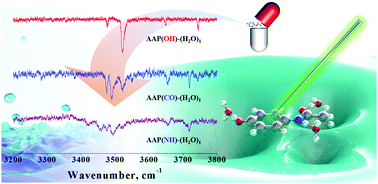
Jet-cooled acetaminophen (AAP)–water clusters, AAP–(H2O)1, were investigated by mass-selected resonant two-photon ionization (R2PI), ultraviolet–ultraviolet hole-burning (UV–UV HB), infrared-dip (IR-dip), and infrared–ultraviolet hole-burning (IR–UV HB) spectroscopy. Each syn- and anti-AAP rotamer has three distinctive binding sites (–OH, >CO, and >NH) for a water molecule, thus 6 different AAP–(H2O)1 conformers are expected to exist in the molecular beam. The origin bands of the AAP(OH)–(H2O)1 and AAP(CO)–(H2O)1 conformers (including their syn- and anti-conformers) in the R2PI spectrum are shifted to red and blue compared to those of the AAP monomer, respectively. These frequency shifts upon complexation between a water molecule and a specific binding site of AAP are also predicted by theoretical calculations. The spectral assignments of the origin bands in the R2PI spectra and the IR vibrational bands in the IR-dip spectra of the four lowest-energy conformers of AAP–(H2O)1, [syn- and anti-AAP(OH)–(H2O)1 and syn- and anti-AAP(CO)–(H2O)1], are aided by ab initio and time-dependent density functional theory (TDDFT) calculations. Further investigation of the IR-dip spectra has revealed a hydrogen-bonded NH stretching mode, supporting the presence of the syn-AAP(NH)–(H2O)1 conformer. Moreover, by employing IR–UV HB spectroscopy, we have reconfirmed the existence of the syn-AAP(NH)–(H2O)1 conformer, which happened to be buried underneath the broad background contributed by the AAP(OH)–(H2O)1 conformers. These observations have led us to conclude that all of the possible conformers of AAP–(H2O)1 have been found in this study.
 Please wait while we load your content...
Please wait while we load your content...

