
In search of the best DFT functional for dealing with organic anionic species†
 *a
*a
Abstract
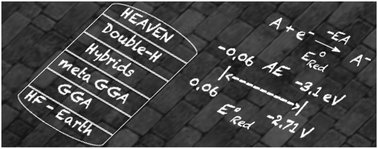
Quantum chemical computational methods are thought to have problems in dealing with unstable organic anions. This work assesses the ability of different Density Functional Theory (DFT) functionals to reproduce the electron affinity and reduction potential of organic compounds. The performance of 23 DFT functionals was evaluated by computing the negative electron affinities (from 0 eV to −3.0 eV) and reduction potentials in acetonitrile (from 0 to −2.7 V). In general, most of the hybrid GGA functionals work fine in the prediction of electron affinities, BPW91, B3PW91 and M06 being the best in each class of functionals (pure, hybrid and meta-GGA functionals, respectively). On the other hand, the ab initio post-Hartree–Fock methods, MP2 and coupled-cluster (CCSD(T)), as well as the double hybrid functionals, B2PLYP and mPW2PLYP, usually fail. For compounds with EAs lower than −1.75 eV, a method for stabilizing the anion, based on solvation with the IEFPCM model, was employed. In this case, BPW91, PBE0 and M06-HF could be the recommended option for the pure, hybrid and meta-GGA functionals, respectively. The situation improves for the evaluation and prediction of redox potentials. In this case the performance of the DFT functionals is better, in part because the solvent assists in the stabilization of the anions. Nevertheless, there is a systematic bias in the calculation of absolute redox potentials, which could be corrected by using a redox partner that helps by the cancellation of errors. In this case, the hybrid and meta-GGA functionals B3PW91, PBE0, TPSSh and M06 are also among the best for computing redox potentials with a mean absolute deviation (MAD) lower than 0.13 V.
 Please wait while we load your content...
Please wait while we load your content...

