
The unique role of bond length in the glassy dynamics of colloidal polymers
Abstract
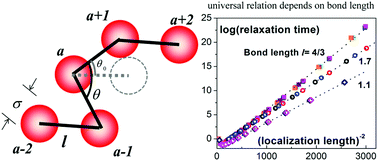
Bond length is generally not considered as a controllable variable for molecular polymers. Hence, no experimental, simulation or theoretical research, to our knowledge, has examined the influence of bond length on the glassy dynamics of polymers. Recently, a new class of assembling materials called “colloidal polymers” has been synthesized. These colloidal polymers have advantages over molecular polymers in the visibility and flexibility of tuning, for example, the size and shape of the “monomers”, the interaction, and the bond length. Dense suspension of colloidal polymers will become a very promising ideal model system for exploring the fundamental problems in the glass transition of chain “molecules”. Here, we study the static structure and activated dynamics of hard-sphere colloidal polymers by generalizing the colloidal nonlinear Langevin equation theory to colloidal polymers. Surprisingly, we find that the bond length plays a critical and unique role in many aspects. For instance, the universal relations of the characteristic local lengths and the activated barrier versus the “degree of supercooling”, and the structural relaxation versus local vibrational motion are found to be dependent on bond length and independent of chain length and rigidity. We hope that our findings inspire future experimental and simulation research studies on the glassy dynamics of colloidal polymers.
 Please wait while we load your content...
Please wait while we load your content...

