
Highly effective sites and selectivity of nitrogen-doped graphene/CNT catalysts for CO2 electrochemical reduction†
Abstract
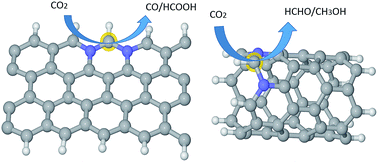
Metal-free catalysts, such as graphene/carbon nanostructures, are highly cost-effective to replace expensive noble metals for CO2 reduction if fundamental issues, such as active sites and selectivity, are clearly understood. Using both density functional theory (DFT) and ab initio molecular dynamic calculations, we show that the interplay of N-doping and curvature can effectively tune the activity and selectivity of graphene/carbon-nanotube (CNT) catalysts. The CO2 activation barrier can be optimized to 0.58 eV for graphitic-N doped graphene edges, compared with 1.3 eV in the un-doped counterpart. The graphene catalyst without curvature shows strong selectivity for CO/HCOOH production, whereas the (6, 0) CNT with a high degree of curvature is effective for both CH3OH and HCHO production. Curvature is also very influential to tune the overpotential for a given product, e.g. from 1.5 to 0.02 V for CO production and from 1.29 to 0.49 V for CH3OH production. Hence, the graphene/CNT nanostructures offer great scope and flexibility for effective tunning of catalyst efficiency and selectivity, as shown here for CO2 reduction.
- This article is part of the themed collections: Global Energy Challenges: Hydrogen Energy, Top 50 Articles of 2016: Energy and Catalysis, Top 50 Articles of 2016: Materials Chemistry and Nanoscience and Top 50 Articles of 2016: Physical Chemistry
 Please wait while we load your content...
Please wait while we load your content...

