
Higher-order structural interrogation of antibodies using middle-down hydrogen/deuterium exchange mass spectrometry†
Abstract
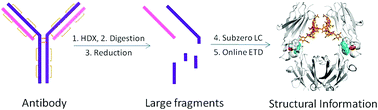
Although X-ray crystallography is the “gold standard” method for protein higher-order structure analysis, the challenges of antibody crystallization and the time-consuming data analysis involved make this technique unsuitable for routine structural studies of antibodies. In addition, crystallography cannot be used for the structural characterization of an antibody in solution, under conditions where antibody drugs are active. Intact antibodies are also too large and too complex for NMR. Top-down mass spectrometry coupled to hydrogen/deuterium exchange (HDX) is a powerful tool for high-resolution protein structural characterization, but its success declines rapidly as protein size increases. Here we report for the first time a new hybrid “middle-down” HDX approach that overcomes this limitation through enabling the nonspecific enzyme pepsin to perform specific restricted digestion at low pH prior to HPLC separation at subzero temperatures and online electron transfer dissociation (ETD). Three large specific peptic fragments (12 to 25 kDa) were observed from the heavy chain and light chain of a therapeutic antibody Herceptin, together with a few smaller fragments from the middle portion of the heavy chain. The average amino-acid resolutions obtained by ETD were around two residues, with single-residue resolution in many regions. This middle-down approach is also applicable to other antibodies. It provided HDX information on the entire light chain, and 95.3% of the heavy chain, representing 96.8% of the entire antibody (150 kDa). The structural effects of glycosylation on Herceptin were determined at close-to-single residue level by this method.
 Please wait while we load your content...
Please wait while we load your content...

