
Quantum mechanical based approaches for predicting pKa values of carboxylic acids: evaluating the performance of different strategies†
 *a
*a
Abstract
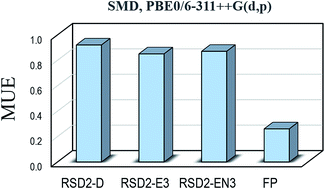
The performance of different computational protocols for predicting pKa values of carboxylic acids, in aqueous solution, has been evaluated by comparison with experimental data. A set of 14 carboxylic acids was used to that purpose, including both aliphatic and aromatic species, as well as molecules of different chemical complexity. Two general strategies were explored, namely the reaction schemes design (RSD) and the fitting parameters (FP) methods. Within RSD, the reaction scheme chosen to represent the deprotonation process is the aspect influencing the most the agreement with the experiments. Other aspects, with modest but not negligible influence, are the way in which the solvation energies are calculated, and the used density functional theory (DFT) approach. The best results within the RSD strategy were found for the EN3 reaction scheme and the PBE0 functional, which led to mean unsigned errors (MUE) and root mean square errors (RMSE) equal to 0.87 and 1.02 pKa units, respectively. However, the FP method over-performs RSD, with MUE and RMSE values below 0.35 pKa units for most of the tested DFT approaches. Accordingly, the FP method is recommended for estimating pKa values of carboxylic acids for which this information is still unknown, especially in combination with the PBE and PBE0 functionals and the SMD solvent model.
 Please wait while we load your content...
Please wait while we load your content...

