
Predicted lithium–iron compounds under high pressure
Abstract
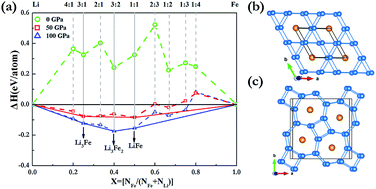
Pressure can induce significant changes in atomic and electronic structures and can in some cases even induce compound formation between elements that do not bond under ambient conditions. Here, we have extensively explored the Li–Fe system at high pressure using the effective CALYPSO algorithm in combination with first-principles calculations. Strikingly, our results show that the stoichiometries of LiFe, Li3Fe and Li3Fe2 have stability regimes on a phase diagram. It is found that both LiFe and Li3Fe2 adopted a fully developed three-dimensional framework. With an increasing Li content, a Li bonding pattern evolves from occupying diamond sublattice to armchair chains, and to the host frameworks constructed by 3- and 6-membered planar rings and 4- and 7-membered planar rings in which linear Fe chains are located in small channels. Our findings put forward a further understanding of the crystal structures and electronic properties of Li–Fe compounds at high pressures.
 Please wait while we load your content...
Please wait while we load your content...

