
Thermoelectric properties of half-Heusler ZrNiPb by using first principles calculations†
Abstract
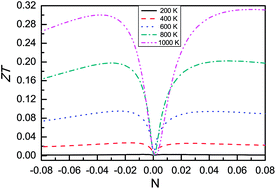
We investigate the electronic structures and thermoelectric properties of a recently synthesized half-Heusler ZrNiPb compound by using a generalized gradient approximation (GGA) and GGA plus spin–orbit coupling (GGA + SOC). The calculated results show that ZrNiPb is an indirect-gap semiconductor. Within the constant scattering time approximation, semi-classic transport coefficients are calculated through solving Boltzmann transport equations. It is found that SOC has a more obvious influence on the power factor in p-type doping than in n-type doping, leading to a reduced effect in p-type doping. These can be explained by considering the SOC influences on the valence bands and the conduction bands near the Fermi level. The lattice thermal conductivity as a function of temperature is calculated, and the corresponding lattice thermal conductivity is 14.5 W m−1 K−1 at room temperature. By comparing the experimental transport coefficients with the calculated ones, the scattering time is obtained as 0.333 × 10−14 s. Finally, the thermoelectric figure of merit ZT can be obtained, and the ZT value can be as high as 0.30 at high temperature by choosing an appropriate doping level. It is possible to reduce the lattice thermal conductivity using point defects and boundaries, and allow half-Heusler ZrNiPb to become a potential parent candidate for efficient thermoelectric materials.
 Please wait while we load your content...
Please wait while we load your content...

