
Vibrational analysis and chemical activity of paracetamol–oxalic acid cocrystal based on monomer and dimer calculations: DFT and AIM approach†
Abstract
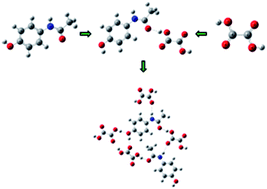
The study of structural and spectral characteristics of a paracetamol–oxalic acid (PRA–OXA) cocrystal has been carried out using two models (monomer and dimer), with the aim to understand the supramolecular structure and intramolecular interactions within the cocrystal. The cocrystal has been characterized by infrared and Raman spectroscopy combined with quantum chemical calculations molecular electrostatic potential surface (MEPS), frontier orbital analysis and electronic reactivity descriptors were used to understand the role of interactions involved in affecting the chemical reactivity of individual molecules in the cocrystal. It is observed that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, N–H and O–H groups of paracetamol are involved in hydrogen bonds to form cocrystals. NBO analysis suggests that the two types of interactions LP(1)(N8) → π*(C9–O10) and LP(2)(O10) → σ*(O25–H28) are responsible for the stability of the molecule. AIM analysis suggested that the non-covalent interactions are moderate in nature. The calculated HOMO–LUMO energies reveal that the charge transfer occurs within the cocrystal. Chemical reactivity parameters show that the cocrystal is more active than paracetamol.
O, N–H and O–H groups of paracetamol are involved in hydrogen bonds to form cocrystals. NBO analysis suggests that the two types of interactions LP(1)(N8) → π*(C9–O10) and LP(2)(O10) → σ*(O25–H28) are responsible for the stability of the molecule. AIM analysis suggested that the non-covalent interactions are moderate in nature. The calculated HOMO–LUMO energies reveal that the charge transfer occurs within the cocrystal. Chemical reactivity parameters show that the cocrystal is more active than paracetamol.
 Please wait while we load your content...
Please wait while we load your content...

