
The evolution of a stereoselective synthesis of the C1–C16 fragment of bryostatins†
Abstract
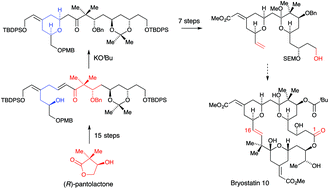
The development of a synthesis of the C1–C16 fragment of bryostatins in which the key step is a stereoselective oxy-Michael reaction used to assemble the cis-2,6-disubstituted tetrahydropyran with the exocyclic alkene already installed, is described. Following early work using Stille reactions to prepare precursors for oxy-Michael reactions, a more efficient route was devised based on a ketophosphonate-aldehyde condensation to prepare the key dienone. Synthesis of the aldehyde required for the ketophosphonate condensation involved the highly stereoselective addition of a diorganocuprate derived from allylmagnesium bromide and copper(I) iodide to the methyl 5-hydroxyhex-2-ynoate prepared by ring-opening of a protected glycidol using lithiated methyl propiolate. Ester reduction, selective alcohol protection and oxidative cleavage of the terminal double bond gave the required aldehyde. The ketophosphonate was prepared in 13 steps from (R)-pantolactone using a synthesis based on a chelation controlled aldol condensation and an anti-selective reduction of a 3-hydroxyketone. Following the ketophosphonate-aldehyde reaction, selective deprotection followed by treatment with base effected the oxy-Michael reaction that gave the cis-2,6-disubstituted tetrahydropyran via thermodynamic control. Subsequent functional group manipulation led to the synthesis of a hydroxy ester that corresponded to the C1–C16 fragment of the bryostatins in 23 steps from (R)-pantolactone. The synthesis was repeated using slightly different protecting groups for a study of a ring-closing metathesis approach to the bryostatins.
 Please wait while we load your content...
Please wait while we load your content...

