
First principles calculations of solid-state thermionic transport in layered van der Waals heterostructures†
Abstract
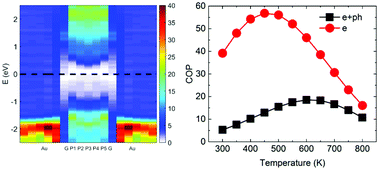
This work aims at understanding solid-state energy conversion and transport in layered (van der Waals) heterostructures in contact with metallic electrodes via a first-principles approach. As an illustration, a graphene/phosphorene/graphene heterostructure in contact with gold electrodes is studied by using density functional theory (DFT)-based first principles calculations combined with real space Green's function (GF) formalism. We show that for a monolayer phosphorene, quantum tunneling dominates the transport. By adding more phosphorene layers, one can switch from tunneling-dominated transport to thermionic-dominated transport, resulting in transporting more heat per charge carrier, thus, enhancing the cooling coefficient of performance. The use of layered van der Waals heterostructures has two advantages: (a) thermionic transport barriers can be tuned by changing the number of layers, and (b) thermal conductance across these non-covalent structures is very weak. The phonon thermal conductance of the present van der Waals heterostructure is found to be 4.1 MW m−2 K−1 which is one order of magnitude lower than the lowest value for that of covalently-bonded interfaces. The thermionic coefficient of performance for the proposed device is 18.5 at 600 K corresponding to an equivalent ZT of 0.13, which is significant for nanoscale devices. This study shows that layered van der Waals structures have great potential to be used as solid-state energy-conversion devices.
 Please wait while we load your content...
Please wait while we load your content...

