
Theoretical characterization of the surface composition of ruthenium nanoparticles in equilibrium with syngas†
Abstract
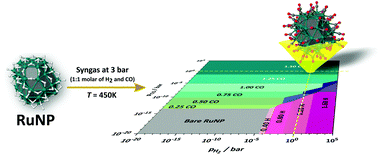
A deeper understanding of the relationship between experimental reaction conditions and the surface composition of nanoparticles is crucial in order to elucidate mechanisms involved in nanocatalysis. In the framework of the Fischer–Tropsch synthesis, a resolution of this complex puzzle requires a detailed understanding of the interaction of CO and H with the surface of the catalyst. In this context, the single- and co-adsorption of CO and H to the surface of a 1 nm ruthenium nanoparticle has been investigated with density functional theory. Using several indexes (d-band center, crystal overlap Hamilton population, density of states), a systematic analysis of the bond properties and of the electronic states has also been done, in order to bring an understanding of structure/property relationships at the nanoscale. The H : CO surface composition of this ruthenium nanoparticle exposed to syngas has been evaluated according to a thermodynamic model fed with DFT energies. Such ab initio thermodynamic calculations give access to the optimal H : CO coverage values under a wide range of experimental conditions, through the construction of free energy phase diagrams. Surprisingly, under the Fischer–Tropsch synthesis experimental conditions, and in agreement with new experiments, only CO species are adsorbed at the surface of the nanoparticle. These findings shed new light on the possible reaction pathways underlying the Fischer–Tropsch synthesis, and specifically the initiation of the reaction. It is finally shown that the joint knowledge of the surface composition and energy descriptors can help to identify possible reaction intermediates.
 Please wait while we load your content...
Please wait while we load your content...

