
Targeting the protein backbone with aryl halides: systematic comparison of halogen bonding and π⋯π interactions using N-methylacetamide†‡
F. M.
Boeckler  *ab
*ab
*ab
Abstract
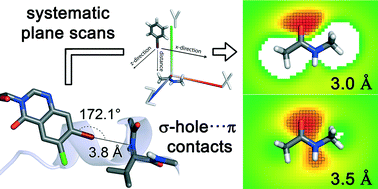
The ubiquitous amide moiety of the protein backbone is an essential interaction partner in any binding site. Using quantum mechanical calculations, we evaluate how to target this moiety through halogen bonding. In contrast to previously employed atom-centric spherical scans, we make use of planar scans to additionally account for the delocalised π-electrons of the amide. The scans showed that perpendicular interaction geometries are moderately strong while favouring the carbonyl oxygen atom at lower distances. Gradually moving to a parallel arrangement results in a transition from σ-hole interactions toward π⋯π and dipolar interactions and higher interaction energies.
 Please wait while we load your content...
Please wait while we load your content...