
Identification of potential CCR5 inhibitors through pharmacophore-based virtual screening, molecular dynamics simulation and binding free energy analysis†
Abstract
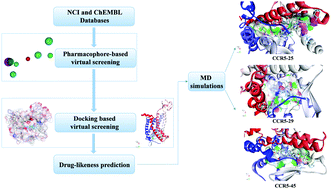
CC chemokine receptor 5 (CCR5), a member of G protein-coupled receptors (GPCRs), plays a vital role in inflammatory responses to infection. Alterations in the expression of CCR5 have been correlated with disease progression in many types of cancers. The idea of using CCR5 as a target for therapeutic intervention has been demonstrated to prevent disease progression. To date, only a few compounds have been reported as CCR5 inhibitors. In this study, a series of CCR5 antagonists were used to construct pharmacophore models. Then the optimal model was utilized as a 3D query to identify novel chemical entities from structural databases. After refinement by molecular docking, drug-likeness analysis, molecular dynamics simulations (MDS) and binding free energy analysis, three potential inhibitors (25, 29 and 45) were identified. MD simulations suggested that the screened compounds retained the important common binding mode known for CCR5 inhibitors (maraviroc and nifeviroc), which occupied the bottom of a pocket and stabilized the conformation of CCR5. During the binding process, van der Waals interactions provided the substantial driving force. The most favorable contributions were from Tyr37, Trp86, Tyr89, Tyr108, Phe109, Phe112, Gln194, Thr195, Ile198, Trp248, Tyr251, Leu255, Thr259, Met279, Glu283 and Met287. The above results suggest that the hybrid strategy would provide a basis for rational drug design.
 Please wait while we load your content...
Please wait while we load your content...