
Energetic and flexibility properties captured by long molecular dynamics simulations of a membrane-embedded pMHCII–TCR complex†
Abstract
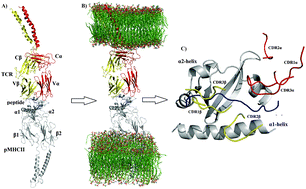
Although crystallographic data have provided important molecular insight into the interactions in the pMHC–TCR complex, the inherent features of this structural approach cause it to only provide a static picture of the interactions. While unbiased molecular dynamics simulations (UMDSs) have provided important information about the dynamic structural behavior of the pMHC–TCR complex, most of them have modeled the pMHC–TCR complex as soluble, when in physiological conditions, this complex is membrane bound; therefore, following this latter UMDS protocol might hamper important dynamic results. In this contribution, we performed three independent 300 ns-long UMDSs of the pMHCII–TCR complex anchored in two opposing membranes to explore the structural and energetic properties of the recognition of pMHCII by the TCR. The conformational ensemble generated through UMDSs was subjected to clustering and Cartesian principal component analyses (cPCA) to explore the dynamical behavior of the pMHCII–TCR association. Furthermore, based on the conformational population sampled through UMDSs, the effective binding free energy, per-residue free energy decomposition, and alanine scanning mutations were explored for the native pMHCII–TCR complex, as well as for 12 mutations (p1–p12MHCII–TCR) introduced in the native peptide. Clustering analyses and cPCA provide insight into the rocking motion of the TCR onto pMHCII, together with the presence of new electrostatic interactions not observed through crystallographic methods. Energetic results provide evidence of the main contributors to the pMHC–TCR complex formation as well as the key residues involved in this molecular recognition process.
 Please wait while we load your content...
Please wait while we load your content...