
Molecular simulation studies on the binding selectivity of 2-anilino-4-(thiazol-5-yl)-pyrimidines in complexes with CDK2 and CDK7†
Abstract
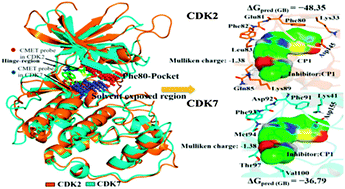
Cyclin dependent kinase 2 (CDK2) was regarded as a potentially therapeutic target for cancer therapy. Since the CDK family includes couples of high homology members, designing CDK2-selective inhibitors would provide valuable opportunities for the development of anticancer drugs with optimal efficacy. In this study, three thiazo-5-yl-pyrimidines as CDK2 inhibitors with different selectivity over cyclin dependent kinase 7 (CDK7) were examined to study the selectivity mechanism using a combined approach of computational techniques of flexible docking, EasyMIFs, molecular electrostatic potential (MESP), natural bond orbital (NBO), molecular dynamics (MD) simulations, and binding free energy calculations. Molecular simulations elicited new chemical insights into steric and electronic complementarities of these molecules to the binding sites of CDK2 and CDK7. The computed binding free energies were consistent with the ranking of their experimental binding affinities on CDK2 and CDK7. We also conducted in silico mutations of three key amino acids (CDK2: Gln85, Lys89, Asp145) to examine their impact on ligand binding with MD simulations and binding free energy calculations. The results indicated that these residues exhibited a strong tendency to mediate ligand–protein interactions through the H-bond and vdW interaction with CDK2-selective inhibitor. The present work may provide a better structural understanding of the molecular mechanism of CDK2 selective inhibition. The new computational insights presented in this study are expected to be valuable for the guidelines and development of new potent CDK2 inhibitors.
- This article is part of the themed collection: Chemical Biology in Molecular BioSystems
 Please wait while we load your content...
Please wait while we load your content...