
Transfer hydrogenation of nitroarenes with hydrazine at near-room temperature catalysed by a MoO2 catalyst†
Abstract
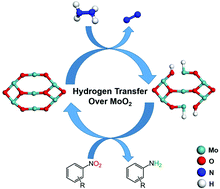
We present an experimental and computational study of the elementary steps of hydrazine hydrogen transfer on crystalline MoO2, and demonstrate its unique bifunctional metallic-basic properties in a catalytic hydrogenation reaction. Density functional theory (DFT) calculations suggest that the stepwise hydrogen transfer via the prior cleavage of the N–H bond rather than the N–N bond, is the key step to create the dissociated hydride and proton species on the dual Mo and O sites, marking its difference with common oxides. Crystalline MoO2 shows exceptionally high chemoselectivity toward the nitro reduction over C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C
C, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, and CN groups at room temperature and lower, down to 0 °C, rendering it as a promising catalytic material for hydrogenation reactions.
C, and CN groups at room temperature and lower, down to 0 °C, rendering it as a promising catalytic material for hydrogenation reactions.
 Please wait while we load your content...
Please wait while we load your content...

