
Electronic and nuclear contributions to time-resolved optical and X-ray absorption spectra of hematite and insights into photoelectrochemical performance†
Abstract
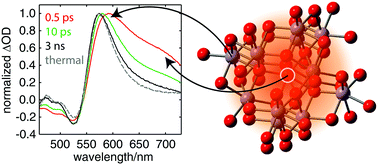
Ultrafast time-resolved studies of photocatalytic thin films can provide a wealth of information crucial for understanding and thereby improving the performance of these materials by directly probing electronic structure, reaction intermediates, and charge carrier dynamics. The interpretation of transient spectra, however, can be complicated by thermally induced structural distortions, which appear within the first few picoseconds following excitation due to carrier–phonon scattering. Here we present a comparison of ex situ steady-state thermal difference spectra and transient absorption spectra spanning from NIR to hard X-ray energies of hematite thin films grown by atomic layer deposition. We find that beyond the first 100 picoseconds, the transient spectra measured for all excitation wavelengths and probe energies are almost entirely due to thermal effects as the lattice expands in response to the ultrafast temperature jump and then cools to room temperature on the microsecond timescale. At earlier times, a broad excited state absorption band that is assigned to free carriers appears at 675 nm, and the lifetime and shape of this feature also appear to be mostly independent of excitation wavelength. The combined spectroscopic data, which are modeled with density functional theory and full multiple scattering calculations, support an assignment of the optical absorption spectrum of hematite that involves two LMCT bands that nearly span the visible spectrum. Our results also suggest a framework for shifting the ligand-to-metal charge transfer absorption bands of ferric oxide films from the near-UV further into the visible part of the solar spectrum to improve solar conversion efficiency.
 Please wait while we load your content...
Please wait while we load your content...

