
Coordination and insertion of alkenes and alkynes in AuIII complexes: nature of the intermediates from a computational perspective†
Abstract
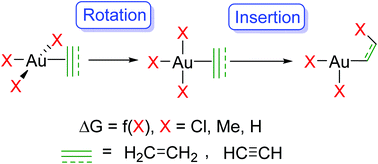
The contribution of AuIII species to catalysis is still debated due to the limited number of characterized intermediates with this oxidation state. In particular, the coordination of alkenes and alkynes to AuIII followed by insertion into AuIII–X bonds has been suggested but rarely proven experimentally. Here, these reactions are explored by means of DFT and CCSD(T) calculations considering [AuX3(L)] and [AuX2(L)2]+ complexes. In these complexes, L = ethylene and acetylene have been chosen as substrates of high interest and representative of any unsaturated organic substrate, whereas X is Cl, Me or H, as found in metal salts and as model for intermediates involved in catalysis. Isoelectronic PtII complexes are also considered for comparison. Ethylene coordination occurs preferentially perpendicular for all X except H, whereas for acetylene, coordination takes place in-plane for all X except Cl. These coordination isomers can represent either minima (intermediates) or saddle points (transition states) on the potential energy surface, depending on X. NBO analysis shows how this variety of structures results from the combination of electronic (M–L donation and back-donation) and steric (cis L–X repulsion) effects. With the sole exception of [AuMe2(ethylene)2]+, rotation of the unsaturated ligand and insertion into a cis Au–X bond involve low to moderate energy barriers, ΔG‡ = 2.5 to 23.5 kcal mol−1, and are thermodynamically feasible, ΔG = 4.3 to −47.2 kcal mol−1. The paucity of experimental observations for such reactions should thus be caused by other factors, like the participation of the intermediates and products in competitive side reactions including the reductive elimination of XCHnCHnX (n = 1 or 2).
- This article is part of the themed collection: Celebrating the 2017 RSC Prize and Award Winners
 Please wait while we load your content...
Please wait while we load your content...

