
First-principles molecular dynamics simulation of the Ca2UO2(CO3)3 complex in water†
Abstract
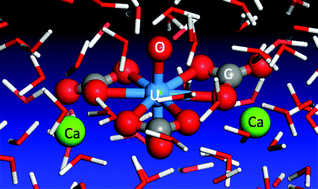
Recent experiments have shown that the neutral Ca2UO2(CO3)3 complex is the dominant species of uranium in many uranyl-containing streams. However, the structure and solvation of such a species in water has not been investigated from first principles. Herein we present a first principles molecular dynamics perspective of the Ca2UO2(CO3)3 complex in water based on density functional theory and Born–Oppenheimer approximation. We find that the Ca2UO2(CO3)3 complex is very stable in our simulation timeframe for three different concentrations considered and that the key distances from our simulation are in good agreement with the experimental data from extended X-ray absorption fine structure (EXAFS) spectroscopy. More important, we find that the two Ca ions bind differently in the complex, as a result of the hydrogen-bonding network around the whole complex. This finding invites confirmation from time-resolved EXAFS and has implications in understanding the dissociative equilibrium of the Ca2UO2(CO3)3 complex in water.
- This article is part of the themed collection: New Talent: Americas
 Please wait while we load your content...
Please wait while we load your content...

