
Investigating the electronic structure of a supported metal nanoparticle: Pd in SiCN

Abstract
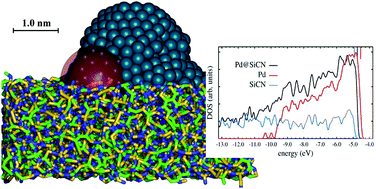
We investigate the electronic structure of a Palladium nanoparticle that is partially embedded in a matrix of silicon carbonitride. From classical molecular dynamics simulations we first obtain a representative atomic structure. This geometry then serves as input to density-functional theory calculations that allow us to access the electronic structure of the combined system of particle and matrix. In order to make the computations feasible, we devise a subsystem strategy for calculating the relevant electronic properties. We analyze the Kohn–Sham density of states and pay particular attention to d-states which are prone to be affected by electronic self-interaction. We find that the density of states close to the Fermi level is dominated by states that originate from the Palladium nanoparticle. The matrix has little direct effect on the electronic structure of the metal. Our results contribute to explaining why silicon carbonitride does not have detrimental effects on the catalytic properties of palladium particles and can serve positively as a stabilizing mechanical support.
 Please wait while we load your content...
Please wait while we load your content...

