
A theoretical study of weak interactions in phenylenediamine homodimer clusters†
Abstract
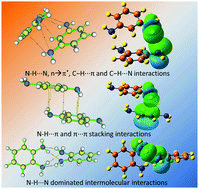
Weak intermolecular interactions in phenylenediamine dimer (pdd) clusters are studied by dispersion-corrected density functional theory (DFT) calculations. Along with the optimization of geometric structures and the calculation of interaction energies, we employ molecular electrostatic potential (MEP) mapping, natural bond orbital (NBO) analysis and quantum theory of atoms in molecule (AIM) to analyze the origin and relative energetic contributions of the weak interactions in these pdd systems. It is revealed that the most stable o-phenylenediamine dimer (opdd) cluster is dominated by N–H⋯N hydrogen bonds, the p-phenylenediamine dimer (ppdd) cluster is largely stabilized by N–H⋯π and π⋯π stacking interactions, while the m-phenylenediamine dimer (mpdd) cluster is mainly held by a combination of n → π*, C–H⋯π and C–H⋯N interactions. Energy decomposition analysis (EDA) of the total interaction energies of these clusters further demonstrates that the weak intermolecular interactions are associated with electrostatic and dispersion contributions. Structural spectroscopic analysis is also addressed depicting the coexistence of multiple intermolecular interactions which give rise to the spectral variation in wavenumbers of the infrared and Raman activities. Insights into the weak interactions of pdds help us to understand the molecular mechanisms involved in biochemistry and self-assembly materials.
 Please wait while we load your content...
Please wait while we load your content...

