
Direct simulation of electron transfer in the cobalt hexammine(ii/iii) self-exchange reaction†
Abstract
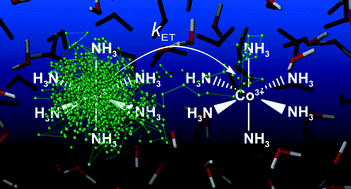
We present an atomistic simulation of the cobalt hexammine(II/III) self-exchange reaction using path integral (PI) methods. We construct a simple force field for the system in its reactant state that includes parameters for both atom–atom interactions, and interactions with an explicit transferring electron represented in the PI framework. We then calculate the outer sphere free energy barrier due to solvent reorganization from a PI molecular dynamics simulation and we obtain the dynamic transmission coefficient using ring polymer molecular dynamics. Combining these calculated values with literature values for the inner sphere reorganization energy, we obtain a reaction rate in good agreement with experimental measurements. The protocol introduced here circumvents the need for complex, system-specific force field parameterization along an assumed reaction coordinate making it sufficiently accurate, efficient, and broadly applicable to the study of both adiabatic and nonadiabatic charge transfer reactions in transition metal complexes.
 Please wait while we load your content...
Please wait while we load your content...

