
Why are sec-alkylperoxyl bimolecular self-reactions orders of magnitude faster than the analogous reactions of tert-alkylperoxyls? The unanticipated role of CH hydrogen bond donation†

Abstract
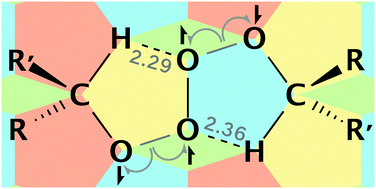
High-level ab initio calculations are used to identify the mechanism of secondary (and primary) alkylperoxyl radical termination and explain why their reactions are much faster than their tertiary counterparts. Contrary to existing literature, the decomposition of both tertiary and non-tertiary tetroxides follows the same asymmetric two-step bond cleavage pathway to form a caged intermediate of overall singlet multiplicity comprising triplet oxygen and two alkoxyl radicals. The alpha hydrogen atoms of non-tertiary species facilitate this process by forming unexpected CH⋯O hydrogen bonds to the evolving O2. For non-tertiary peroxyls, subsequent alpha hydrogen atom transfer then yields the experimentally observed non-radical products, ketone, alcohol and O2, whereas for tertiary species, this reaction is precluded and cage escape of the (unpaired) alkoxyl radicals is a likely outcome with important consequences for autoxidation.
 Please wait while we load your content...
Please wait while we load your content...

