
CoP for hydrogen evolution: implications from hydrogen adsorption†
Abstract
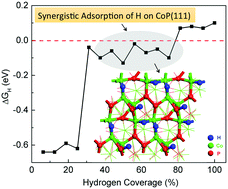
Cobalt phosphide (CoP) is one of the most promising, earth-abundant electrocatalysts discovered to date for hydrogen evolution reaction (HER), yet the mechanism is not well understood. Since hydrogen adsorption is a key factor of HER activity, here we examine the adsorption of atomic hydrogen on the low-Miller-index surfaces of CoP, including (111), (110), (100), and (011), by using periodic density functional theory. From the calculated Gibbs free energy of adsorption, we predict that (111), (110), and (011) surfaces will have good catalytic activities for HER. From ab initio atomistic thermodynamics, we find that the stabilities of the surfaces at 1 atm H2 and 300 K follow the trend of (111) > (100) ∼ (110) ≫ (011). On the most stable (111) surface, both Co bridge sites and P top sites are found to be able to adsorb hydrogen with a close-to-zero free energy change and the synergy of proximal Co and P atoms on the surface results in a better HER activity. Our work provides important insights into CoP's excellent HER activity and a basis for further mechanistic understanding of HER on CoP and other transition-metal phosphides.
 Please wait while we load your content...
Please wait while we load your content...

