
π–π stacking between polyaromatic hydrocarbon sheets beyond dispersion interactions†
Abstract
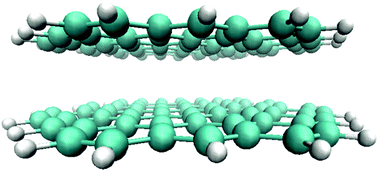
High level ab initio calculations ranging from coupled cluster methods including explicitly correlated approaches to standard second order Møller–Plesset theory using spin scaling (SOS-MP2) have been performed on sandwich and slipped parallel dimer structures of a series of quasi one-dimensional acenes and on two-dimensional sheets containing the pyrene to coronene series encircled with two layers of benzene rings. Sandwich (graphitic AA type) and slipped parallel (AB type) structures were considered and, within the given symmetry restrictions, full geometry optimizations were performed. Basis set superposition effects have been considered. The computed geometries show a significant biconcave deviation of the two-dimensional sheets from planarity with the central intersheet C⋯C distances considerably smaller that van der Waals distances. The computed intersheet binding energy per carbon atom extrapolated for N → ∞ of −74.3 meV (1.713 kcal mol−1) per atom agrees quite well with an experimental defoliation energy of −52 meV (1.199 kcal mol−1) per atom (−67 meV (1.545 kcal mol−1) per carbon atom without corrections for H binding contributions) for polyaromatic hydrocarbons (PAHs) from graphite. A limited investigation of density functional theory (DFT) calculations using empirical dispersion contributions has been performed also showing a significant underbinding character of the D3 method. For most of the DFT variants investigated the graphene sheet models retain a quasi-planar structure in strong contrast to the aforementioned SOS-MP2 results.
 Please wait while we load your content...
Please wait while we load your content...

