
Ethylene decomposition on Ir(111): initial path to graphene formation†
Abstract
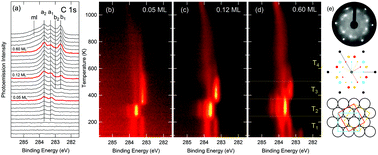
The complete mechanism behind the thermal decomposition of ethylene (C2H4) on Ir(111), which is the first step of graphene growth, is established for the first time employing a combination of experimental and theoretical methods. High-resolution X-ray photoelectron spectroscopy was employed, along with calculations of core level binding-energies, to identify the surface species and their evolution as the surface temperature is increased. To understand the experimental results, we have developed a reaction sequence between the various CnHm species, from ethylene to C monomers and dimers, based on ab initio density functional calculations of all the energy barriers and the Arrhenius prefactors for the most important processes. The resulting temperature evolution of all species obtained from the simulated kinetics of ethylene decomposition agrees with photoemission measurements. The molecular dissociation mechanism begins with the dehydrogenation of ethylene to vinylidene (CH2C), which is then converted to acetylene (CHCH) by the removal and addition of an H atom. The C–C bond is then broken to form methylidyne (CH), and in the same temperature range a small amount of ethylidyne (CH3C) is produced. Finally methylidyne dehydrogenates to produce C monomers that are available for the early stage nucleation of the graphene islands.
 Please wait while we load your content...
Please wait while we load your content...

