
Molecular mechanisms responsible for hydrate anti-agglomerant performance†
Abstract
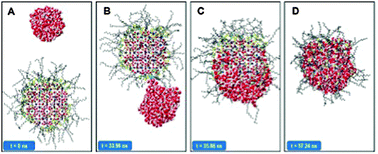
Steered and equilibrium molecular dynamics simulations were employed to study the coalescence of a sI hydrate particle and a water droplet within a hydrocarbon mixture. The size of both the hydrate particle and the water droplet is comparable to that of the aqueous core in reverse micelles. The simulations were repeated in the presence of various quaternary ammonium chloride surfactants. We investigated the effects due to different groups on the quaternary head group (e.g. methyl vs. butyl groups), as well as different hydrophobic tail lengths (e.g. n-hexadecyl vs. n-dodecyl tails) on the surfactants' ability to prevent coalescence. Visual inspection of sequences of simulation snapshots indicates that when the water droplet is not covered by surfactants it is more likely to approach the hydrate particle, penetrate the protective surfactant film, reach the hydrate surface, and coalesce with the hydrate than when surfactants are present on both surfaces. Force–distance profiles obtained from steered molecular dynamics simulations and free energy profiles obtained from umbrella sampling suggest that surfactants with butyl tripods on the quaternary head group and hydrophobic tails with size similar to the solvent molecules can act as effective anti-agglomerants. These results qualitatively agree with macroscopic experimental observations. The simulation results provide additional insights, which could be useful in flow assurance applications: the butyl tripod provides adhesion between surfactants and hydrates; when the length of the surfactant tail is compatible with that of the hydrocarbon in the liquid phase a protective film can form on the hydrate; however, once a molecularly thin chain of water molecules forms through the anti-agglomerant film, connecting the water droplet and the hydrate, water flows to the hydrate and coalescence is inevitable.
 Please wait while we load your content...
Please wait while we load your content...

