
A comparative study of small 3d-metal oxide (FeO)n, (CoO)n, and (NiO)n clusters
Abstract
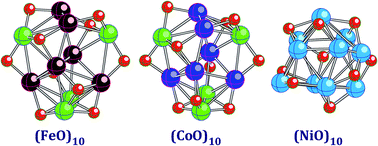
Geometrical and electronic structures of the 3d-metal oxide clusters (FeO)n, (CoO)n, and (NiO)n are computed using density functional theory with the generalized gradient approximation in the range of 1 ≤ n ≤ 10. It is found that the cluster geometries are similar in the (FeO)n and (CoO)n series but noticeably different in the (NiO)n series for several values of n. All of the lowest total energy states are found to possess relatively small spin multiplicities and are either antiferromagnetic or ferrimagnetic except for the states of (NiO)3, (NiO)4, (NiO)9, and (NiO)10, which are ferromagnetic. The computed polarizabilities per atom undergo a steep decrease when compared to the atomic values of the MO monomers (M = Fe, Co, and Ni). Surprisingly, the polarizability does not strongly depend on either M or n in all the considered series when n varies from 3 to 10. The binding energies per atom are the largest in the (FeO)n series, followed by the binding energies of (CoO)n and (NiO)n.
 Please wait while we load your content...
Please wait while we load your content...

