
A first-principles study of pressure-induced phase transformation in a rare-earth formate framework†
Abstract
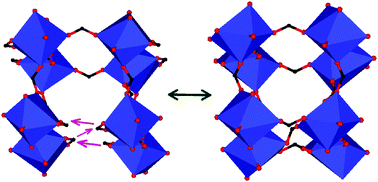
Among the panoply of exciting properties that metal–organic frameworks (MOFs) exhibit, fully reversible pressure-induced phase transformations (PIPTs) are particularly interesting as they intrinsically relate to the flexibility of MOFs. Recently, a number of MOFs have been reported to exhibit this feature, which is attributed to bond rearrangement with applied pressure. However, the experimental assessment of whether a given MOF exhibits PIPT or not requires sophisticated instruments as well as detailed structural investigations. Can we capture such low pressure transformations through simulations is the question we seek to answer in this paper. For this, we have performed first-principles calculations based on the density functional theory, on a MOF, [tmenH2][Y(HCOO)4]2 (tmenH22+ = N,N,N′,N′-tetramethylethylenediammonium). The estimated lattice constants for both the parent and product phases of the PIPT agree well with the earlier experimental results available for the same MOF with erbium. Importantly, the results confirm the observed PIPT, and thus provide theoretical corroborative evidence for the experimental findings. Our calculations offer insights into the energetics involved and reveal that the less dense phase is energetically more stable than the denser phase. From detailed analyses of the two phases, we correlate the changes in bonding and electronic structure across the PIPT with elastic and electronic conduction behavior that can be verified experimentally, to develop a deeper understanding of the PIPT in MOFs.
- This article is part of the themed collection: In celebration of Tony Cheetham’s 70th birthday
 Please wait while we load your content...
Please wait while we load your content...

