
A close examination of the structure and dynamics of HC(NH2)2PbI3 by MD simulations and group theory†

Abstract
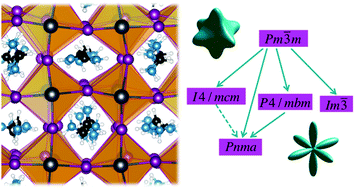
The formamidinium lead iodide hybrid perovskite is studied using first principles molecular dynamics simulations and further analyzed using group theory. The simulations are performed on large supercells containing 768 atoms under isothermal and fully anisotropic isobaric conditions. Two trajectories, one at 300 K and another at 450 K, were extended for over 50 ps in order to perform a detailed assessment of the rotational dynamics of organic cations. The characteristic rotations of the cation are analyzed by defining two rotation axes. It is found that the formamidinium molecules rotate preferentially around the direction parallel to the line connecting the two nitrogen atoms. The rotational dynamics shows some characteristics already observed in methylammonium lead iodide, like the heterogeneous dynamics at room temperature that disappears at 450 K. The orientational probability of the molecules is explored in terms of an expansion in cubic harmonics up to the 12th order. It reveals a strong directionality at room temperature that relaxes when increasing the temperature. These findings are further rationalized using Landau and group theories suggesting a mixed displacive/order–disorder structural instability at lower temperatures.
- This article is part of the themed collection: Physical chemistry of hybrid perovskite solar cells
 Please wait while we load your content...
Please wait while we load your content...

